

TEXTO DO CANAL BRASIL SEM ALERGIA NO YOUTUBE
O texto pode conter erros na transcrição uma vez que são geradas automaticamente pelo YouTube. Não deixe de assistir o vídeo em nosso canal.
0:00 Oi boa noite a todos 0:03 e eu vou me apresentar vocês 0:06 Olá meu nome é Luís Berger Fronteira 0:09 outro médico eu sou pega de pela 0:13 Universidade Federal de Medicina em 0:17 medicina e só tem um pós-doutorado Em 0:21 genética pela pela Fiocruz Rio de 0:25 Janeiro eu tinha um serviço de 0:28 imunologia e alergia 30 spray Natal da 0:31 Santa Casa do Rio de Janeiro e 0:34 provavelmente esse serviço o serviço 0:36 mais antigo do Brasil foi criado pelo 0:41 que eu sou Clementino Fraga filho e seu 0:43 primeiro chefe tem eu fui Assistente por 0:47 isso eu sou Antonio de Oliveira Lima em 0:50 o nosso nosso 0:54 e a nossa palestra eu resolvi fazê-la em 0:59 português e vai ser tem o objetivo de 1:03 transmitir para vocês os conceitos 1:06 básicos de e mundial do Cristo permanece 1:10 Norte eu vou falando em português mas eu 1:14 vou fazer uma saudação ao produtor 1:16 Jackson Black é em inglês e o Gabriel 1:19 depois eu volto para o português eu 1:23 quero agradecer muito a organização você 1:28 inventa protagonista na Carvalho e o 1:31 Doutor Marcelo popular 1:34 é o dia do impressor Jackson André isso 1:38 aproveite o Norte do Rio Words about 1:41 Christmas on 1:43 Oi Espera ai ai cursei Christian and 1:47 triggering Factor of motion 1:51 e está a Nice place in the chapter of 1:54 life 1:55 a entrar ai ajuda de admitir e ouvir sua 2:00 opção 2:01 A Lenda de líquidos thank you and Doctor 2:06 good download for Action and citation 2:11 eu não aguento tu ser mais fim eu peguei 2:16 a amar mais pó 2:19 a montando o inglês mas temos hoje a 2:22 rede honra modera um seminário 2:24 internacional de ciências farmacêutica a 2:27 perspectiva diagnóstica e terapêutica 2:30 utilizando a tecnologia Christopher a 2:34 os jogadores desse evento é a faculdade 2:43 de Filosofia Ciências e Letras do Alto 2:45 São Francisco o professor Thales Renato 2:48 Ferreira Carvalho e Canadá Gene therapy 2:52 on 2:54 nós não temos os palestrantes professor 2:56 Jackson indo é porque eu sou do 2:58 departamento de medicina molecular Vila 3:00 Walter Becker Canadá ele para falar 3:04 sobre o uso do cristo para tratar 3:06 doenças hereditárias no segundo o 3:09 palestrante será o Doutor Gabriel o amor 3:11 EA do trabalho do laboratório de 3:13 medicina molecular da Universidade laval 3:16 Quebec Canadá ar e falará sobre o uso 3:24 e a uso da tecnologia derivada do cristo 3:27 para detectar infecções virais e eu como 3:32 moderador 3:33 e o apoio departamento de medicina 3:36 molecular da Universidade laval laval 3:39 Quebec Canadá 3:41 E a faculdade pelos dias sem acento em 3:43 letras do Alto São Francisco Associação 3:47 Brasileira beneficente de apoio Alerta e 3:50 pernas grades e no teto e Associação de 3:53 saúde sustentabilidade a Deus que é uma 3:55 associação Nossa com objetivos sociais 3:59 na medicina no serviço de imunologia 4:02 Clínica e alergia da Santa Casa do Rio 4:05 de Janeiro e a clínica imunoderm ó eu 4:09 vou começar a falar com vocês são 4:13 assuntos tentando explicar de maneira 4:17 bem pragmática e os conceitos 4:21 fundamental do Cristo atrás 9 4:24 bom então o sistema aquífero slide 4:27 inglês Sweet Life Into Space on with it 4:32 e é são repetições pane Drone eu vou 4:36 falar para vocês também que que 4:38 significa essa palavra pamidronico ou 4:40 seja repetições põe crônicas curtas 4:43 agrupadas e regularmente interespaçadas 4:47 consiste em pequenas porções DNA 4:50 bacteriano composta por reflexões no 4:54 Clothes não não DNA bacteriano e no 4:57 nosso também depois foi descoberto mas 4:59 tenho nós temos dessas repetições curtas 5:03 e ir essa descoberta do Cristo ela foi 5:08 começou a ser falada Tá indo em 1986 em 5:14 2010 começou a aparecer as pesquisas 5:18 sobre o Cristo e o nosso serviço nosso 5:21 serviço de imunologia da Santa Casa um 5:24 acordo de cooperação cientista nós 5:26 começamos a trabalhar com isso há cinco 5:29 anos e hoje nós temos junto com 5:31 professor da questão do é e a Fiocruz no 5:37 Rio de Janeiro nós estamos fazendo uma 5:40 uma reflexão de uma técnica utiliza do 5:43 curso opção para detecção muito mais 5:50 rápido muito mais barato por vírus 5:52 inclusive do navio dos piratas da 5:57 posição nós estamos aguardando também a 6:00 vinda bem breve do Gabriel lamont que é 6:02 um dos Mundos 6:03 A vaga é participar dessa repetição 6:11 dessa técnica do Cristo feita pelo 6:13 professor Jackson André Então o sistema 6:16 Cristo pois ele é o significa no nosso 6:22 DNA são reflexões curtas e agrupadas 6:26 regularmente na porção do DNA bacteriano 6:30 compostos de reflexões nucleotídicas 6:33 vamos até que vocês vão entender embaixo 6:35 pega uma dessas reflexões encontra-se 6:39 adjacente a um próprio espaçador 6:42 espaçador de DNA e corresponde a uma 6:46 região não purificada e ferida no DNA 6:49 bacteriano após o contato com genomas 6:53 vivas dores provenientes de vírus 6:56 bacteriófago bacteriófago e transmitir 6:59 Ou seja a detecção deste conjunto crista 7:03 quase 7:03 a receita é uma bactéria e se defende da 7:08 invasão dos bacteriófagos e é muito 7:11 interessante eu gosto de dizer que só 7:13 Deus mesmo explicaria isso a bactéria 7:16 então no seu DNA ele tem essa esse 7:19 conjunto de repetições de nucleotídeos é 7:25 e ele é capaz de induzir a formação de 7:32 uma enzima que as nozes e vai lizar o 7:37 DNA o DNA do vírus e incorpora esse 7:42 pedaço de DNA nessa essa parte de DNA ou 7:48 RNA no seu próprio DNA então eles serão 7:52 os próprios espaçadores e eles ficam 7:55 localizado adjacente a esse a esse 7:59 Cristo da bactéria 8:01 e na próxima vez essa bactéria entrar em 8:04 contacto com o vírus então o DNA dela é 8:08 capaz de detectar essa esse próprio 8:11 espaçador que tá no DNA e e que a 8:14 sequência de base por dentro do 8:17 hidrogenada do vidro e com isso ela sabe 8:19 que tá sendo agredida já tem uma memória 8:22 e ela então o DNA dela ela induz a 8:25 formação de uma rede a e vai produzir 8:29 uma enzima para realizar o vírus e as em 8:34 cima é o caso dos é muito interessante e 8:37 esse pros espaçador que é um pedaço do 8:41 RNA ou DNA do vírus ele ele não tem 8:43 penetrância por isso que é uma feijão 8:46 não pode ficante do lado dele que fica a 8:50 sequência do Cristian 8:53 E aí 8:55 e a transcrição do Bloco do pisca 8:59 resulte em pequenos fragmentos de RNA 9:01 com capacidade de desenhar o 9:04 reconhecimento de um DNA Episódio 9:07 específico e atuar com um guia de modo a 9:10 orientar anunciado caspases irá promover 9:15 a free Fire linguagem e consequentemente 9:17 eliminado DNA e vazou caso esse entre 9:21 novamente em contato com a bactéria 9:23 atuando como importante mecanismo de 9:25 defesa. DNA sem Lagoas a justamente isso 9:28 quando o princípio e tem essa repetição 9:33 de base nitrogenada e está do Lago do 9:39 RNA ou DNA depois trazido pela Caixa do 9:45 lisado pela cidade nova incorporada no 9:48 DNA da bactéria quando o vírus entra na 9:52 bactéria essa bactéria então 9:55 do do kriska ido do fragmento do RNA ou 10:00 DNA do vírus ele detecta o vírus produz 10:04 RNA e esse ganhar todas as fases 9 que 10:09 vai é destruir e subir 10:14 e o Cristian então ele é um repetições 10:19 palindrômica vou explicar vocês né 10:22 porque é isso é de pequena repetições 10:26 quando eletrônica opção em três passadas 10:30 regularmente pode tem um desenho que eu 10:32 vou explicar o Cristo é uma família de 10:36 sequência de DNA base de clonadas 10:40 encontrada no organismo procariontes do 10:43 DNA em bactérias e na área essa essa 10:48 algum tempo atrás da chave as pensar que 10:51 a porção até mais tarde que não é uma 10:54 bactéria e é muito interessante que é o 10:56 a estrutura mais antiga encontrada na 10:59 terra 11:00 o e as sequências é bons são derivados 11:05 do DNA do vírus Então essa sequência de 11:10 bases nitrogenadas que tá do lado do 11:13 Cristã ela é derivada do bacteriófago no 11:16 sector essa bactéria 11:19 bom então Cristo é capaz de detectar uma 11:22 nova invasão e capaz de produzir um caso 11:26 9 11:29 e na Idade então o Creeper sistema 11:32 crítica é um sistema imune da da matéria 11:35 do Povo carioca e confere resistência a 11:40 elementos genéticos estranho e assim 11:42 apresenta dentro do se apresenta dentro 11:45 do Anísio ou dos Fábio e fornece então 11:49 uma forma de unidade adquirida essa 11:52 bactéria 11:56 bom então quê que é uma sequência 11:58 palindromica essa essa sequência com 12:01 eletrônica ela só não existe nos isso 12:04 somente nas bactérias Não essa esse 12:07 monitor hidrônico também serve para 12:10 determinar as palavras e tem um pão 12:12 hidrônico é uma palavra ou sequência de 12:16 aminoácidos jornadas e você lendo do 12:20 início do fim ou do tempo o início é a 12:22 mesma coisa então isso aí quando você 12:25 tem o nome de uma pessoa Ana esse nome é 12:27 Tony dromico tanto faz duas ele do do 12:30 sufixo prefixo E você tem o mesmo nome e 12:34 é justamente isso que acontece nas 12:37 sequências Clone Drone cá do DNA vocês 12:40 podem ver aqui e você tem do da parte do 12:44 5 para o três linhas ou dos cinco para o 12:49 todinha eu tenho a sequência pequena de 12:52 bases nitrogenadas que elas estão G 12:56 e se e a outra gatt1 a mesma coisa tanto 13:00 faz então com isso mas você ler esse 13:03 custo esse conjunto de pequenos bases 13:07 nitrogenadas da esquerda para direita ou 13:09 da direita para esquerda essa sequência 13:12 existe também no ser humano ela fica no 13:15 DNA e do lado dela e fica aquela 13:20 sequência na bactéria e fica do lado do 13:23 PIS Portela no DNA dela aquela sequência 13:26 de bases nitrogenadas do vírus e com 13:30 isso onde esse vírus e essa sequência é 13:33 muito importante que essa sequência de 13:35 bases nitrogenadas do vírus ela não tem 13:38 que prestar atenção nessa internet 13:39 falando duas proteínas nas duas dentre 13:42 Mas ela não se ela não tem atividade 13:44 genética então o espelho e junto com é a 13:49 só sequência do vírus 13:52 e quem atividade genética que vai 13:54 detectar a nova invasão viral e será 13:57 capaz então de produzirem temas que vão 14:00 destruir o vírus outro a sequência 14:03 correta tônica que você vê a ser a e 14:06 aqui você tem a a a aqui também e te GT 14:13 e na outra na outra a faixa ttgc então 14:18 eu tanto faz eu eu ligo daqui para cá ou 14:21 daqui para lá então isso que é o que 14:22 significa o nome panetone então 14:27 terminando que o objetivo não sei se eu 14:30 estou sendo Claro é da para vocês os 14:32 fundamentos básicos essenciais da do 14:36 significado de Cristo hidratação 14:38 bacteriana sobre o vírus é com seus 14:40 postos entender tanto a a palestra do 14:44 professor Jackson Blair e opressores 14:46 aqui também eu quero deixar claro que 14:49 ele foi um dos primeiros pesquisadores 14:50 do mundo a trabalhar 14:52 E aí 14:53 o lilac eu vi também ei mandei para para 14:57 a universidade de laval na para o 15:02 laboratório dele ou Nossa coordenação 15:05 científica atual pessoais das luz ele 15:09 ficou um pelo lá na laboratório Tô tendo 15:12 é é aprendendo essa reta de utilização 15:17 do pista como terapia para você vir aqui 15:20 você tem a membrana celular da bactéria 15:22 e sempre bacteriófago ele essa tem o DNA 15:26 viral compadre a dupla e a Aqui tá o DNA 15:30 da bactéria Aqui nós temos a série A 15:33 Série crise quando a bactéria o DNA dela 15:35 ela vai produzir com as pazes e vai 15:40 utilizar especificamente aquilo que dá 15:44 ela a capacidade de colocar do lado do 15:49 da série crise cânceres esse conjunto de 15:52 base campeonatos 15:53 a ganhar do Viral isso que eu disse para 15:56 você que para vocês que a sensação aqui 15:59 é uma ação Milagrosa né como é que se 16:03 explica que ela consegue através das 16:06 caspases quebrar o DNA do vírus 16:11 especificamente trazer Para incorporar o 16:13 CD ao DNA dela essa sequência fiz um 16:16 vírus ela é que faz esse espaço né ela 16:20 não tem penetrância genética como esse 16:22 vir entrar virus entrar novamente vai 16:24 acontecer o DNA dele é sensibilizar a 16:28 bactéria ela tem aqui esse Prisma é que 16:33 tem aquela sequência que se repete tanto 16:35 da esquerda para direita e eu posso ler 16:37 mas aqui tem importante a gente colocou 16:39 no seu DNA é a sequência de bases do 16:42 Leonardo do vírus então estudar ela 16:45 agora uma mulher memória adquirida pela 16:47 não é um memory naquela foi adquirida 16:49 entrando as férias em contato com o piso 16:51 então com isso ela vai te acho 16:53 e vai produzir o dinheiro vai conduzir o 16:56 rma e vai produzir o com o complexo de 16:59 trás pago e as pás novas e vai 17:02 direcionar o tiro existe não ia ouvir 17:05 bom então isso é é o tentei espero que 17:11 eu tenha sido Claro na explicação desse 17:14 mecanismo do crisma e dos conceitos 17:17 fundamentais depois nós vamos abrir a 17:21 pergunta se eles podem perguntar e agora 17:23 eu vou voltar aperto atenção para você 17:26 só voltar a falar inglês só para para 17:28 começar o iniciar não chamar o professor 17:33 o Léo Professor Jackson Black aí o canal 17:36 a distant or Speech Welcome to this 17:40 symposium






 O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em
O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em  O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em
O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em 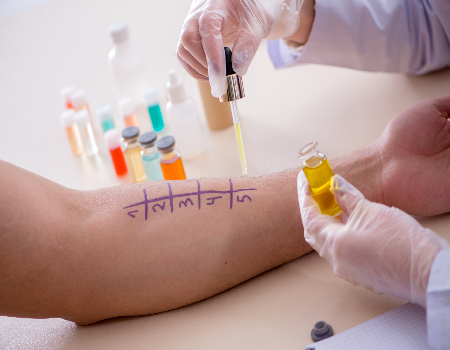 O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em
O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em  O atendimento especializado do Brasil Sem Alergia foca no diagnóstico preciso e no tratamento acessível de patologias como dermatite atópica, asma 🌬️, bronquite 🫁, rinite e prurigo estrófulo (alergia a picadas de insetos 🦟). Nossa nova unidade no Campo Limpo, em São Paulo, segue o padrão de excelência da rede, oferecendo testes para detectar alergias causadas por poeira, pelo de animais e alimentos. Como um projeto de inclusão social que complementa o
O atendimento especializado do Brasil Sem Alergia foca no diagnóstico preciso e no tratamento acessível de patologias como dermatite atópica, asma 🌬️, bronquite 🫁, rinite e prurigo estrófulo (alergia a picadas de insetos 🦟). Nossa nova unidade no Campo Limpo, em São Paulo, segue o padrão de excelência da rede, oferecendo testes para detectar alergias causadas por poeira, pelo de animais e alimentos. Como um projeto de inclusão social que complementa o 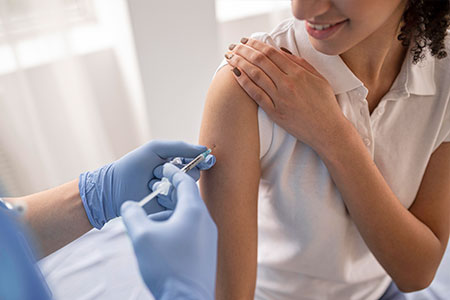 Proteja-se com Vacinas no Projeto Brasil Sem Alergia!As vacinas são uma forma segura e eficaz de prevenir doenças como gripe , febre amarela 🟡, meningite , pneumonia 🫁 e diversas alergias! O Projeto Brasil Sem Alergia oferece algumas vacinas e medicamentos gratuitos, em campanhas e com patrocínio, para ajudar você a ter mais saúde e qualidade de vida.Nossas UnidadesRio de Janeiro:
Proteja-se com Vacinas no Projeto Brasil Sem Alergia!As vacinas são uma forma segura e eficaz de prevenir doenças como gripe , febre amarela 🟡, meningite , pneumonia 🫁 e diversas alergias! O Projeto Brasil Sem Alergia oferece algumas vacinas e medicamentos gratuitos, em campanhas e com patrocínio, para ajudar você a ter mais saúde e qualidade de vida.Nossas UnidadesRio de Janeiro:  Para esta variação, o foco é a narrativa histórica e humanizada, detalhando a origem voluntária do projeto na Baixada Fluminense e sua evolução até a chegada à capital paulista. O texto está em formato corrido, otimizado para SEO e com a inclusão da unidade Campo Limpo e seus respectivos contatos.O Projeto Brasil Sem Alergia 🇧🇷❤️ é o resultado de uma trajetória de cuidado e inclusão iniciada em 2007, quando o Dr. Marcello Bossois, médico
Para esta variação, o foco é a narrativa histórica e humanizada, detalhando a origem voluntária do projeto na Baixada Fluminense e sua evolução até a chegada à capital paulista. O texto está em formato corrido, otimizado para SEO e com a inclusão da unidade Campo Limpo e seus respectivos contatos.O Projeto Brasil Sem Alergia 🇧🇷❤️ é o resultado de uma trajetória de cuidado e inclusão iniciada em 2007, quando o Dr. Marcello Bossois, médico 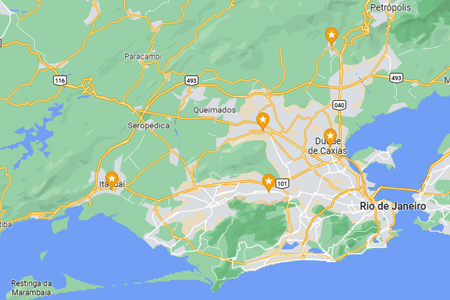 O Projeto Brasil Sem Alergia consolida sua presença nacional e expande seu alcance com unidades fixas no
O Projeto Brasil Sem Alergia consolida sua presença nacional e expande seu alcance com unidades fixas no  O Projeto Brasil Sem Alergia reforça sua missão de inclusão social através de sua unidade móvel e itinerante, que percorre diversas regiões para levar saúde e bem-estar diretamente até a sua comunidade. Com o objetivo de democratizar o acesso ao diagnóstico especializado, nossa clínica móvel oferece consulta com
O Projeto Brasil Sem Alergia reforça sua missão de inclusão social através de sua unidade móvel e itinerante, que percorre diversas regiões para levar saúde e bem-estar diretamente até a sua comunidade. Com o objetivo de democratizar o acesso ao diagnóstico especializado, nossa clínica móvel oferece consulta com 

