

TEXTO DO CANAL BRASIL SEM ALERGIA NO YOUTUBE
O texto pode conter erros na transcrição uma vez que são geradas automaticamente pelo YouTube. Não deixe de assistir o vídeo em nosso canal.
0:28 welcome thank you for having me uh let's see if 0:34 we can get it started hi my name is gabriel lamot and today 0:41 i'll be discussing the detection of cyrus kobe yeah that's much better is the sound 0:47 better now b2 using crispr technology oh perfect namely the test that we've been developing in dr tremblay's laboratory 0:55 studies have confirmed that many individuals infected with the sars kobe 2 virus are asymptomatic carriers of the virus 1:02 this is problematic in that the pandemic might be growing without adequate supervision simply focusing the testing on 1:08 individuals with symptoms as opposed to entire populations is not enough 1:14 to better contain the virus until a large scale distribution of an effective vaccine is underway large scale and 1:20 continuous testing is mandatory the current diagnostic pipeline for 1:26 detecting sars cov2 is a three-step process the first step is to obtain nasopharyngeal swabs from 1:32 patients the next step is to isolate the total rna from said samples and provide the rna required for this 1:40 third step which is to use real-time quantitative vcr machines the rna is first reverse transcribed 1:46 into dna and then amplified a dye is added to the amplification reaction to signal the presence of dna 1:52 strands after a certain number of amplification cycles a positive result will give 1:57 a strong enough signal that the machine flags it as containing the original virus now this process is problematic for a 2:04 few reasons the first is that real-time pcr machines are extremely expensive 2:10 most can range from 15 000 us dollars to well over 90 000 us dollars 2:16 given that it's a fairly specialized piece of equipment and that not all molecular biology laboratories need one 2:22 some get along just fine with a regular pcr machine which costs a much more reasonable 5000 us dollars 2:28 as such widespread testing for the coronifiers using these machines isn't ideal 2:33 acquiring the large number of machines required for widespread testing is a feat in and of itself 2:39 and is likely one of the contributing factors for the sometimes long wait times for results 2:45 many reports have stated that it can take 3 to 24 hours or more to obtain results in a clinical 2:51 diagnostic laboratory over here in canada ontario reports wait times of up to four days to get results 2:58 high-density metropolitan areas like new york are also subject to long lines for testing and then a time to 3:05 results of about seven days for some clinics in that city the creation of new types of detection tests 3:12 is therefore an important step in curbing the spread of this virus an ideal test needs to follow the world 3:19 health organization's criteria for assured that is the test needs to be affordable 3:25 for as many people at risk of infection as possible tests that require extremely expensive reagents or machinery 3:31 are not ideal the test must also be specific to avoid false negatives that way anyone 3:37 who could potentially spread the virus can be identified and put into quarantine the test must be sensitive 3:43 so that few people who don't have the virus are mislabeled as having it and treated improperly 3:48 the test must also be user friendly that is it must be simple to perform and it must be rapid and robust that is 3:55 it must give a time to results that is quick enough to make the most use from the results situations where it can take up to seven 4:02 days to obtain results are clearly far from ideal as by the time the patient receives 4:07 the information they might have infected many others the test must be equipment free as much 4:13 as possible tests that require too many rare and expensive machines aren't ideal for many 4:18 countries finally the test must be deliverable to those who need it 4:24 the discovery of cast 13 or c2 c2 was revolutionary in the field of molecular detection 4:31 this class 2 type 6 crispr effector is similar to the protein cast 9 off of 4:36 which base ending and prime ending were designed this protein is also a member of the crispr family 4:43 unlike cas9 however cast 13 can be programmed to target rnas as opposed to dnas 4:49 by binding to its crispr rna cas13 is primed to target extremely specific rna sequences after locating a target 4:57 sequence the cast 13 protein is activated this causes the protein to begin to cleave 5:03 everything its surroundings in an event called collateral cleavage it cleaves its original target 5:08 and then moves on to start cleaving all surrounding rnas researchers realized that this 5:14 collateral cleavage could be used to turn the protein into a switch for a detection test once 5:19 the target molecule is present the switch is flipped the cast cas13 activated and all surrounding rnas are degraded by 5:27 tagging certain rnas in the solution with a fluorescent molecule and a quencher 5:32 it would therefore become possible to resolve a signal researchers therefore started to develop 5:38 a detection test based on this protein for viruses bacteria and human diseases such as cancer 5:45 however they soon noted that there were certain limitations to the protein and that is limited detection was far from sensitive 5:51 enough for their needs recently certain tests have been designed using only cast 13 5:57 however these tests are less sensitive and tend to use a different cast 13 protein 6:03 researchers at the broad institute decided to supplement the protein with a few others that would amplify the 6:08 target rna to this end dr zhang and his team created a new test called specific high sensitivity 6:16 enzymatic reporter unlocking or sherlock for short this test is based 6:21 on the reverse transcription of target rna into dna afterwards the dna is amplified at a 6:27 constant temperature by using a process called recombinase polymerase amplification 6:32 this technique amplifies a small amount of dna at a constant temperature unlike the more commonly used bcr 6:40 the incorporation of this step also means that the ensuing detection test can target dna since the rpa reaction 6:47 simply amplifies a determined section of dna using two primers the initial genetic sequence can either 6:53 be rna or dna so long as reverse transcriptase enzymes are present for rna 7:00 the amplified sequences are then transcribed using a t7 rna polymerase to create vast quantities of the target 7:07 rna this then allows the cas13 to have a far larger pool of target molecules 7:12 which can activate the enzyme once the enzyme is active it does what we discussed before 7:18 and starts to cleave all surrounding rnas including reporter rnas which give off the easily detectable signal 7:24 which makes the test work in their initial version of the sherlock test two different reporter 7:31 molecules were used the first used a fluorescent molecule as well as a quencher 7:37 and it was able to produce a lot of fluorescence after cleavage of the rna the other 7:44 was designed to interact with a few antibodies and when it was cleaved it appeared as a distinct 7:50 second band on a strip that was quite similar to a pregnancy test 7:55 while this test was good in many ways it had certain drawbacks at the beginning of the pandemic we attempted to replicate it and 8:01 immediately had a few issues the biggest issue by far was that some of the materials required for the test 8:07 to work were only available in extremely limited supply the kits required to perform rpa 8:12 reactions for example took approximately two months to arrive and even then we only received enough 8:18 materials to perform 200 tests this clearly wasn't going to be enough to produce sufficient tests in the 8:24 future additionally the reporter that used a pregnancy style strip was remarkable and seemed to be very 8:31 useful however since it was too difficult to acquire insufficient quantities 8:36 we decided not to use it and to instead focus our efforts on using the fluorescent style reporter rna since the original version 8:44 of the test could not be used by us in the way that it was intended we decided to redesign it 8:51 in redesigning the test we had several options and routes that we could take we knew that we wanted to keep on using 8:57 cast 13 because we had a large supply of this enzyme due to an ongoing collaboration with dr 9:02 alain ghani's lab we also knew that we wanted at all costs to maintain the isothermal nature of 9:08 this test one of the biggest advantages to sherlock was that it didn't require large amounts of expensive equipment 9:14 simply by using a heating block or something of the sort it was possible to get a visual readout 9:20 we therefore decided to employ loop-mediated isothermal amplification or lamp for short this amplification 9:27 strategy was fairly well documented and easy enough to perform based on the use of six primers this 9:33 technique could rapidly amplify large amounts of target dna unlike both rpa and pcr however 9:39 lamp doesn't create identical amplicons of a discrete size instead the six primers can interact to 9:46 create far larger molecules of a repeating sequence initially it was a little difficult to 9:52 incorporate this isothermal amplification strategy into our test as you might remember the step following 9:58 rpa in the original sherlock test was one that produced lots and lots of target rna 10:03 this production of target rna was only possible because the rpa step added a tag that let the t7 rna 10:10 polymerase bind to the dna and begin transcribing it however no one had seemingly ever done 10:16 that with lamp and since this new technique used six primers two of which looped back on themselves to create 10:22 weird dumbbell like structure structures it was difficult to figure out exactly where to incorporate this tag 10:29 after a while we decided to include the tag right in the middle of one of our looping primers this has consistently 10:35 given us very strong results we were therefore able to retrofit the original sherlock 10:41 test with a new amplification strategy all the while still benefiting from cast 13's specificity and signaling 10:48 the new test is therefore using rt lamp as opposed to rt rpa of course 10:54 should we become interested in targeting dna in the future for either certain types of viruses or 11:00 certain bacteria this test would be readily applicable to that as well we also decided to use a fluorescent 11:07 reporter rna instead of the pregnancy style one that we spoke about previously because it seemed to be the most readily 11:13 available material since it doesn't take any particularly advanced machinery to make it work 11:18 we were happy to choose this one 11:24 now that you know how the test works from a theoretical standpoint let's discuss it from a more practical point of view for now 11:31 the test requires that we start with rna that was extracted from a virus using a commercially available kit one 11:37 microliter of this solution is transferred over to the tube containing the rt lamp reaction 11:42 ideally in the future we would like to modify the test so that it becomes possible to directly add a patient's saliva 11:48 to it and continue on from there that would noticeably cut down on the labor required to obtain each reading 11:54 cut down on the overall cost and make it much faster for the results to come in 11:59 after transferring one microliter of the rna extract to the first tube the tube must then be incubated at 65 12:05 degrees celsius for 30 minutes after that one microliter is transferred from the first tube 12:11 into the second which contains the t7 and cast 13 solution after tapping the tube a few times to 12:18 mix it and microcentrifuge it down for a few seconds all you need to do is incubate that tube at 37 degrees celsius 12:25 for 30 minutes at that point all of the reactions are done and that the results are ready to be 12:31 read by exposing the tubes to a wavelength of approximately 490 nanometers 12:36 it's possible to easily detect which samples contain cyrus cov2 those ones will fluoresce a bright green 12:43 those that didn't contain any of the viral specimens of interest will be completely colorless and indistinguishable from a tube filled 12:50 with a little water i'll show you that later now it's worth noting that the dna and rna sequences 12:57 that we have used in this first version of our test were all previously published by different groups 13:03 as such we do not believe that they will result in a positive signal when in the presence of a relevant rna 13:10 this is particularly important because your saliva is full of rna the extraction process for the virus 13:16 does indeed indeed result in viral rnas being captured but there tends to be a whole lot more 13:22 human and bacterial rnas too in addition other coronaviruses that are 13:27 much less dangerous are fairly common in the human population if our tests gave positive signals every time someone came in 13:34 with a common cold then it wouldn't be very effective as such we have based ourselves on 13:39 previously described templates to try and avoid this as much as possible 13:45 in designing this test we tried to make sure that it wasn't overly reliant on complicated machines 13:50 we wanted the entire test to be easy to perform using only a few common laboratory apparatuses 13:56 as a result with this current iteration of the test all we need is one machine that can heat 14:02 pcr-style tubes to 65 degrees celsius and one that can heat 14:07 the pcr tubes to 37 degrees celsius we also need two either p2 or p10 14:14 pipettes for reasons we'll get into later along with their associated peptide tips 14:20 a small microcentrifuge capable of taking pcr tubes is also required just to make sure that the reaction 14:26 mixtures don't touch the top of the tube when you're opening them and potentially contaminating 14:31 all of the surrounding tubes finally a machine capable of giving off ultraviolet wavelengths is necessary 14:38 in this picture you'll see the machine i've been using it's most commonly used to visualize gels in a molecular biology 14:44 lab but it does a really good job of visualizing our positive samples in our test for the two incubation 14:52 periods several different options are available i've been using an old thermocycler today 14:57 as i find it does a really good job of evenly heating my tubes it's worth noting here that this means 15:03 that even old and run-down thermocyclers that no longer do a very good job of cycling between temperatures 15:09 can be readily used in this test unlike the current rtq pcr gold standard that requires 15:16 specialized real-time thermocyclers here any old machine can be used 15:22 it's also possible to use other common laboratory apparatuses such as block heaters that have an 15:27 adapter for pcr tubes please note that in this picture the block heater has an adapter for 1.5 15:34 millimeter microcentrifuge tubes so the true adapter will look a little different 15:39 i've also used incubators to great success even bacterial incubators that operate at a steady 37 degrees 15:46 celsius can perfectly suit the needs of this test so long as the tubes are readily exposed 15:51 to the ambient temperature then positive signals should have no difficulty in resolving 15:56 please note however that when using incubators it's better to have the tubes exposed to the air 16:02 that way they don't waste time as their rack slowly begins to heat up 16:07 water baths are one final option that you can choose personally i tend to avoid this option 16:13 because i find the risk for cross-contamination between samples to be much greater and therefore unideal 16:19 however i have tried to perform rt lamp reactions with this system and so long as you're 16:24 careful it's very possible to do it 16:30 once the reactions have been completed it's possible to use various different machines to visualize the tubes depending on the 16:36 materials your laboratory has personally my machine of choice is a gel uv emitter 16:42 these machines which are more commonly used to visualize agarose gels do a very good job of safely showing 16:48 which tubes are fluorescent and which aren't black lights can also be used 16:53 for this test however i have found them to be slightly less desirable since the reporter rna best absorbs 16:59 wavelengths of 490 nanometers i found that black lights which produce wavelengths of 390 nanometers 17:06 to be less favorable for this test for them to work it's better to visualize the tubes in a dark room 17:13 in both cases the machine should really be placed in a darker room away from excessive light a little 17:19 background light seems to have less of an effect in the case of the gel uv light however 17:25 this picture seen on the left was taken using a generic iphone when the tubes were placed on the gel uv 17:31 light the lights in the room were only partially turned off resulting in lots of ambient lighting 17:37 even if there were no lights directly pointed at the machine given the strong and even radiation that the tubes 17:44 received on this machine it's easy to determine which samples were positive and which were negative 17:50 in the case of the black light on the right it's clear which samples are positive and which are negative 17:55 of course however the fluorescence is not even and it's a little bit harder to detect 18:01 by the naked eye in this case the camera on the iphone took a very good picture 18:06 and made it easier to visualize the tubes overall now there are several important 18:11 considerations to be had when using this test for it to work properly and not give either false positives or false 18:17 negatives it's important to first designate proper working stations for each section of the test it's also 18:24 necessary to follow the predetermined reaction times without varying otherwise the results might start to 18:30 differ slightly finally those handling the test must be careful not to contaminate any of the 18:36 tubes with rnases the first consideration to separate 18:41 working areas is extremely important because lamb is excessively sensitive this technique is known to produce lots 18:48 and lots of concentrated target dna we as well as others have noted that 18:54 when we open tubes that completed the lamp amplification in the same area that we originally used 18:59 to set up the reaction it's possible to contaminate the working area with aerosols containing the 19:04 product if you continue to set up the lamp reactions in those areas afterwards then you run the risk of 19:11 contaminating your new tubes with amplicons from the previous reactions by touching your desk or 19:16 various other contaminated surfaces with your gloves you run the risk of artificially seeding your reaction 19:22 mixtures that contain patient rna with no sarisco v2 and therefore producing positive results 19:29 when there really shouldn't have been any by separating the areas in which you set up the lamp reactions 19:34 and those in which you use the amplified sequences you run less of a risk for this to happen it's 19:40 also necessary to have designated lab coats pipettes and other such materials for each working station 19:47 if those in workstation 2 become contaminated you don't want that coming back into workstation 1. 19:53 please note that this is much less stringent when bringing materials from workstation 1 to workstation 2. 19:59 since it's the rt lamp reaction that runs the greatest risk of contamination bring material for example gloves 20:06 from that station to the next should not be a problem provided that the gloves are still 20:12 relatively clean please note that this situation of contamination via aerosol 20:18 is of much less importance during the second stage of the test as we discussed previously the cast 13 20:24 protein we are using requires a relatively large amount of rna to be functional 20:30 at the present time we are not concerned in the least then aerosol contamination from lamp 20:35 might produce false positives of our cast 13 reactions as we have yet to see a single situation 20:41 where this has happened the second consideration of note is that 20:47 the reaction time must be followed a small discrepancy can be tolerated that is one minute more or less doesn't 20:54 really seem to have a large impact on the test however more than that can start to become problematic 21:01 in the case of the rt lamp reaction external teams as well as us have seen that by doubling the reaction 21:07 time for the lamp false positives can arise as you can see here in both pictures 21:13 a successful lamp reaction can also be visualized on an agarose gel the successful reactions are 21:19 characterized by the presence of several large bands of varying sizes however when the lamp 21:25 reactions go on for too long it's possible to begin amplifying samples with nothing in them 21:30 for example on the left you'll see a gel that was taken from an article in which at 30 minutes the wells 21:37 containing reactions with 10 copies of the target are viral rna and no copies of the 21:42 target viral rna had no bands in the case of the gel on the right a gel that i made three of the wells 21:50 contained reactions that had no target rna to speak of only the n fragment well contained 21:56 target rna at 30 minutes these negative controls did not demonstrate any amplification 22:02 as we expected however at 60 minutes both the published gel and mine started 22:08 to give amplified products in their case the ten and zero copies both started to be amplified 22:15 and in my case one of my negative samples started to get amplified i've performed 22:22 several experiments so far with 30 minute incubation times and so far have never gotten false positives that's 22:29 not to say that it's entirely impossible simply that it's very very unlikely as a 22:35 side note this is a good time to mention that changing gloves frequently when performing the first reaction 22:41 is a good way to avoid these kind of unlikely false positives after prolonged use gloves might become 22:47 contaminated with small amounts of viral rna and that could potentially lead to false positives even if we're only incubating 22:54 at 30 minutes the final consideration to look out for 22:59 is the presence of rnases for those of you who might not be familiar rnases are enzymes that cleave 23:05 all rnas they come across both steps for this test are sensitive to rnases 23:11 the initial arty lamp begins its amplification off of rna so the presence of rnases could be very 23:17 detrimental and therefore lead to false negatives the second step of the test the t7 and cast 13 step uses 23:25 an rna reporter to signal positive tubes if while you're using the tests you start to introduce rnases 23:31 then you run the risk of getting false positives rnases are extremely difficult to remove 23:37 once they're introduced into a system so the best means of protecting your test is by using prevention 23:43 since your hands produce lots of rnases it's mandatory to use gloves at all times when working with the test 23:49 in doing so you will prevent your body's natural rnases from degrading the initial star's cov2 23:54 rna and protect the fluorescent reporter at the end gloves must also be changed whenever 24:00 they come into contact with skin hair or other frequently manipulated objects 24:06 such as doorknobs and personal devices the use of rnas and dnase free 24:12 filtered pipettes is also important for this test by using these kinds of specialized tips 24:19 you can make sure that any possible contaminations you might have in your pipettes doesn't have a chance 24:24 to contaminate your test in areas where the risk for rnas contamination is fairly high 24:30 it might be worthwhile to treat all surfaces or tools with an rnas removal solution these 24:36 kinds of solutions can degrade rnases that might otherwise stick to your gloves and contaminate your samples 24:42 please note i haven't had to use the solution very often so long as you maintain a clean 24:48 working schedule you should be fine the second component for the test 24:54 the one containing t7 and cast 13 is also designed to be able to tolerate a 24:59 certain level of contaminating rnases there is an rnas inhibitor added to the reaction mixture 25:06 to prevent the degradation of the reporter in case the original sars kobe 2 rna 25:11 wasn't completely clean or in case contaminations occurred while you were preparing the tests 25:18 that said the presence of excessive rnases can still fully activate the test seen 25:24 here on the right one microliter of very concentrated rnase a is capable of causing the test tube to 25:30 become fluorescent it's therefore important to take the 25:40 required 25:54 cabrillo 26:02 test that can be widely distributed while also meeting this limit of detection is fully functional for curbing the 26:10 spread of this pandemic while rt-qpcr is much more sensitive 26:15 than this its improved limited detection might not necessarily be that beneficial 26:20 since this technique can pick up rnas freed by infected and dead cells after the infection has 26:26 run its course this form of screening can cause individuals who are nominally sars kovi to rna positive but aren't 26:34 actually infectious to be isolated unnecessarily as you can see here on our left our 26:41 detection test is able to detect samples that have 100 copies per microliter as was stipulated by the model a more 26:49 recent comparison between the different primers that we've been using for lamp has demonstrated that one version of our 26:55 test can detect samples containing only 80 copies per microliter while the other can only detect samples 27:01 containing 1 000. moving forward we're likely to focus our efforts on the first primer set to ensure that 27:08 we meet the limit of detection that was stipulated by the model 27:13 as a final note we're happy to report that our test works not only on fragments of the cyrus 27:19 cov2 genome it also works when exposed to the virus's full rna profile 27:25 initially we began the development of our test by using fragments of the viruses genes that we had 27:31 transcribed ourselves in the lab recently we've used the test on actual 27:36 sars cov2 viruses and have obtained the expected results as you can see on the left 27:42 the first section of the reaction the rt lamp reaction works just as expected on the viral rna 27:48 the top left wells that were run on the agarose gel clearly demonstrate a strong amplification below them three 27:56 reactions were performed with cells that had not been cultured with the virus the viral rnas were created by first 28:02 isolating the viruses from a patient these viruses were then cultured in viroe6 cells 28:08 and after a few days the culture medium was treated to extract any and all rnas that were present the 28:15 viruses were therefore destroyed and their rna was obtained when the lab amplicons that had 28:21 amplified the real virus were transferred over to the second stage of the test they gave the expected 28:26 strong fluorescent signals at the moment what remains to be done is to use our tests on samples 28:33 that have been confirmed positive or negative by rtq pcr the current gold standard in sarsko v2 28:40 detection and see how our test compares this will give us a better indication 28:45 of what our tests sensitivity and specificity are just to wrap up 28:52 i would like to thank dr gary cobinger dr ellen gianni and dr ghibwave for having helped to 28:58 provide finances or materials for this project i'd also like to thank dr tremblay and everyone in his team 29:04 for having helped provide certain insights as i was redesigning this test finally i'd like to thank dr marcelo for 29:12 having helped to organize this entire meeting and the canadian institute for health research 29:17 and the frqs for having helped finance me as i pursued this project 29:27 okay so that was my presentation uh yeah 29:34 thank you gabriel uh i'll take the time just to make a uh to summarize it the best i can here 29:41 for the non-english speakers thank you very much for the presentation thank you okay






 O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em
O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em  O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em
O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em 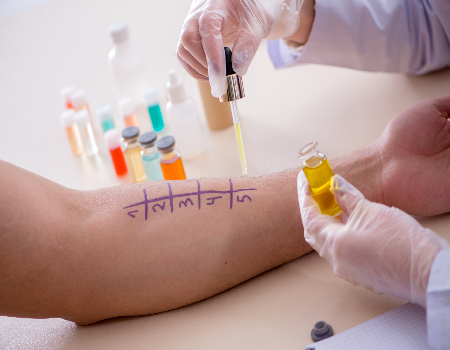 O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em
O Projeto Brasil Sem Alergia consolidou sua trajetória de cuidado e inclusão social em 2007, quando os médicos alergistas e imunologistas Dr. Marcello Bossois e Dra. Patrícia Schlinkert iniciaram um trabalho voluntário em  O atendimento especializado do Brasil Sem Alergia foca no diagnóstico preciso e no tratamento acessível de patologias como dermatite atópica, asma 🌬️, bronquite 🫁, rinite e prurigo estrófulo (alergia a picadas de insetos 🦟). Nossa nova unidade no Campo Limpo, em São Paulo, segue o padrão de excelência da rede, oferecendo testes para detectar alergias causadas por poeira, pelo de animais e alimentos. Como um projeto de inclusão social que complementa o
O atendimento especializado do Brasil Sem Alergia foca no diagnóstico preciso e no tratamento acessível de patologias como dermatite atópica, asma 🌬️, bronquite 🫁, rinite e prurigo estrófulo (alergia a picadas de insetos 🦟). Nossa nova unidade no Campo Limpo, em São Paulo, segue o padrão de excelência da rede, oferecendo testes para detectar alergias causadas por poeira, pelo de animais e alimentos. Como um projeto de inclusão social que complementa o 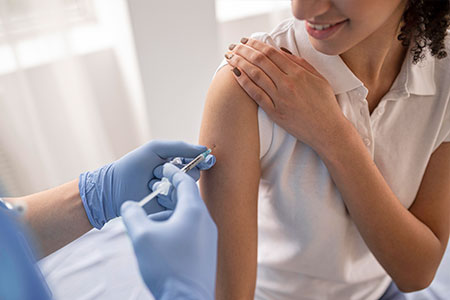 Proteja-se com Vacinas no Projeto Brasil Sem Alergia!As vacinas são uma forma segura e eficaz de prevenir doenças como gripe , febre amarela 🟡, meningite , pneumonia 🫁 e diversas alergias! O Projeto Brasil Sem Alergia oferece algumas vacinas e medicamentos gratuitos, em campanhas e com patrocínio, para ajudar você a ter mais saúde e qualidade de vida.Nossas UnidadesRio de Janeiro:
Proteja-se com Vacinas no Projeto Brasil Sem Alergia!As vacinas são uma forma segura e eficaz de prevenir doenças como gripe , febre amarela 🟡, meningite , pneumonia 🫁 e diversas alergias! O Projeto Brasil Sem Alergia oferece algumas vacinas e medicamentos gratuitos, em campanhas e com patrocínio, para ajudar você a ter mais saúde e qualidade de vida.Nossas UnidadesRio de Janeiro:  Para esta variação, o foco é a narrativa histórica e humanizada, detalhando a origem voluntária do projeto na Baixada Fluminense e sua evolução até a chegada à capital paulista. O texto está em formato corrido, otimizado para SEO e com a inclusão da unidade Campo Limpo e seus respectivos contatos.O Projeto Brasil Sem Alergia 🇧🇷❤️ é o resultado de uma trajetória de cuidado e inclusão iniciada em 2007, quando o Dr. Marcello Bossois, médico
Para esta variação, o foco é a narrativa histórica e humanizada, detalhando a origem voluntária do projeto na Baixada Fluminense e sua evolução até a chegada à capital paulista. O texto está em formato corrido, otimizado para SEO e com a inclusão da unidade Campo Limpo e seus respectivos contatos.O Projeto Brasil Sem Alergia 🇧🇷❤️ é o resultado de uma trajetória de cuidado e inclusão iniciada em 2007, quando o Dr. Marcello Bossois, médico 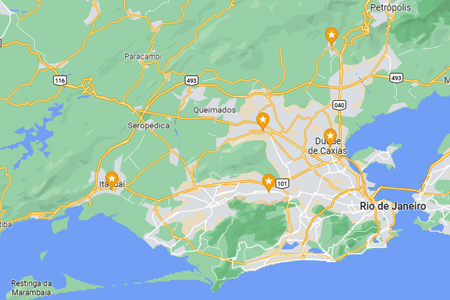 O Projeto Brasil Sem Alergia consolida sua presença nacional e expande seu alcance com unidades fixas no
O Projeto Brasil Sem Alergia consolida sua presença nacional e expande seu alcance com unidades fixas no  O Projeto Brasil Sem Alergia reforça sua missão de inclusão social através de sua unidade móvel e itinerante, que percorre diversas regiões para levar saúde e bem-estar diretamente até a sua comunidade. Com o objetivo de democratizar o acesso ao diagnóstico especializado, nossa clínica móvel oferece consulta com
O Projeto Brasil Sem Alergia reforça sua missão de inclusão social através de sua unidade móvel e itinerante, que percorre diversas regiões para levar saúde e bem-estar diretamente até a sua comunidade. Com o objetivo de democratizar o acesso ao diagnóstico especializado, nossa clínica móvel oferece consulta com 

